BARTweb help page
BARTweb is an interactive web server to predict functional transcriptional regulators that regulate a given gene list or associate with a genomic profile. It is a web interface to BART Python package.
If you use BARTweb in your data analysis or publish the results of BARTweb, please cite the following papers in the main text of your manuscript:
- Wenjing Ma, Zhenjia Wang, Yifan Zhang, Neal E. Magee, Yayi Feng, Ruoyao Shi, Yang Chen, Chongzhi Zang. BARTweb: a web server for transcriptional regulator association analysis. NAR Genomics and Bioinformatics, (2021). DOI: 10.1093/nargab/lqab022
- Zhenjia Wang, Mete Civelek, Clint Miller, Nathan Sheffield, Michael J. Guertin, Chongzhi Zang. BART: a transcription factor prediction tool with query gene sets or epigenomic profiles. Bioinformatics, 34, 2867–2869 (2018). DOI: 10.1093/bioinformatics/bty194
Tutorial Video
Manual
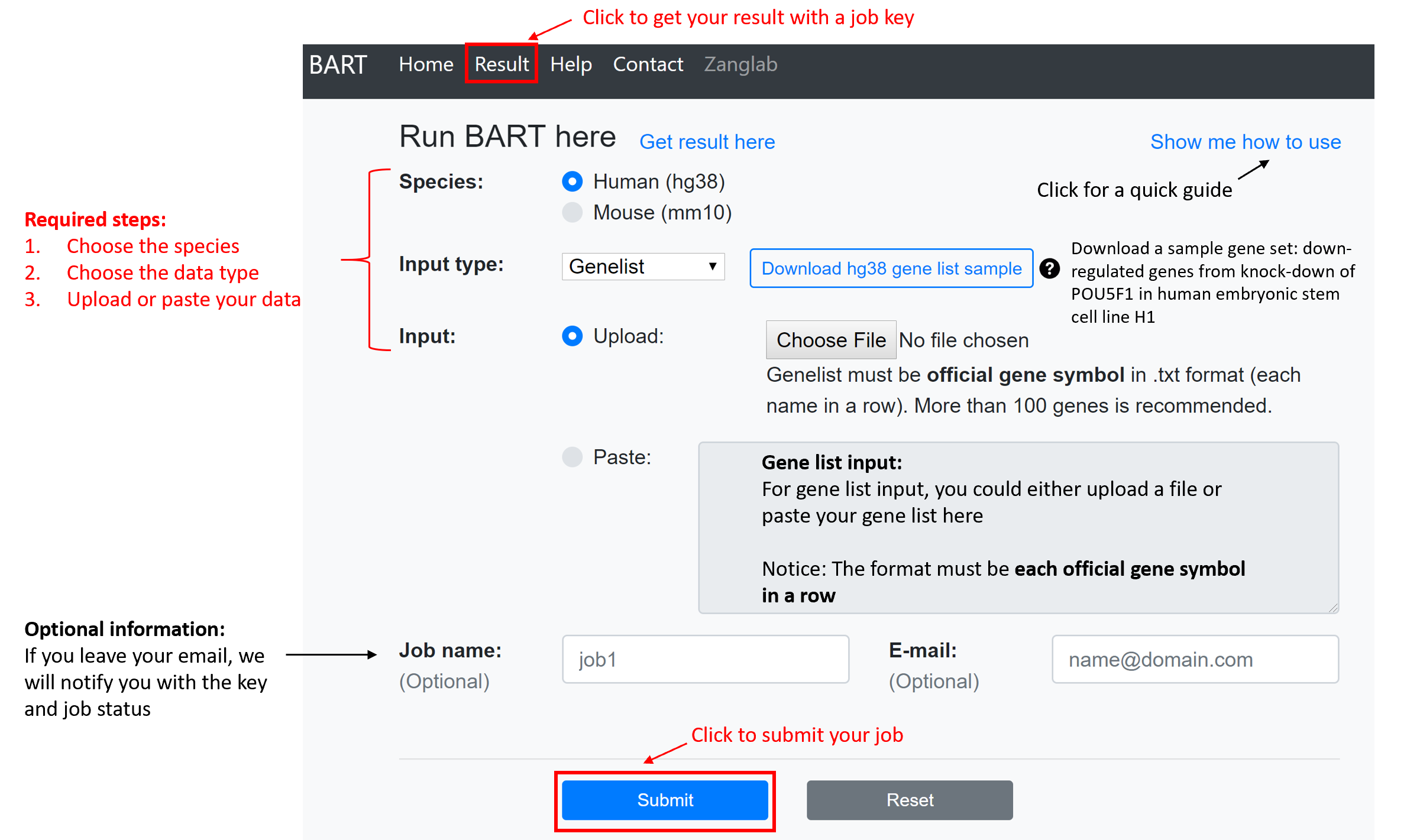
Submit a job
The first three options (species, data type and input data) are required.
Species: Currently, only human (hg38) and mouse (mm10) genomes are supported.
Input data type:
- Gene list: A gene list has to be official gene symbols (HGNC for human or MGI for mouse) in text format. BARTweb can identify functional Transcription Regulators (TRs) that regulate the gene set. At least 100 genes are recommended in the input.
- ChIP-seq data: ChIP-seq data can be mapped reads in either BAM or BED format. BARTweb can identify TRs have a binding profile correlated with a ChIP-seq profile. At least 1 million reads are recommended in the input.
- Region (under BETA testing): The region input has to be a BED file with scored, non-overlapping intervals, for example, ChIP-seq peaks. The scores have to be on 5th row of the BED file. Please make sure that the ChIP-seq data or scored bed file are mapped to the correct genome version (hg38 or mm10). BARTweb can identify TRs enriched in a genomic region set.
If an email address is provided, BARTweb will send email notifications with the job key, job status, and the result URL.
After clicking the “Submit” button, BARTweb will redirect to the result page.
Result page
It usually takes a few minutes to run a job. You can click the “Result” button on the navigation bar or use the job key to get the result or the job status anytime. If the job has not finished, you will see the “still-running” status in the title and a processing log presented. You can click the “Copy Key” button to copy the job key to the clipboard and keep it for your record. If the job fails, the detailed error message will be shown in the processing log. If you cannot figure out possible causes of the error, you are welcome to contact us with the job key for help.
You can share your BART analysis result with others by providing them with the result link URL or the job key. Anyone with a result URL or a job key can open and view the BART analysis result at the user’s own risk.
For successfully finished jobs, the analysis result will be stored on the web server and can be retrieved with the job key for 180 days after the job submission.
How to interpret the BART result
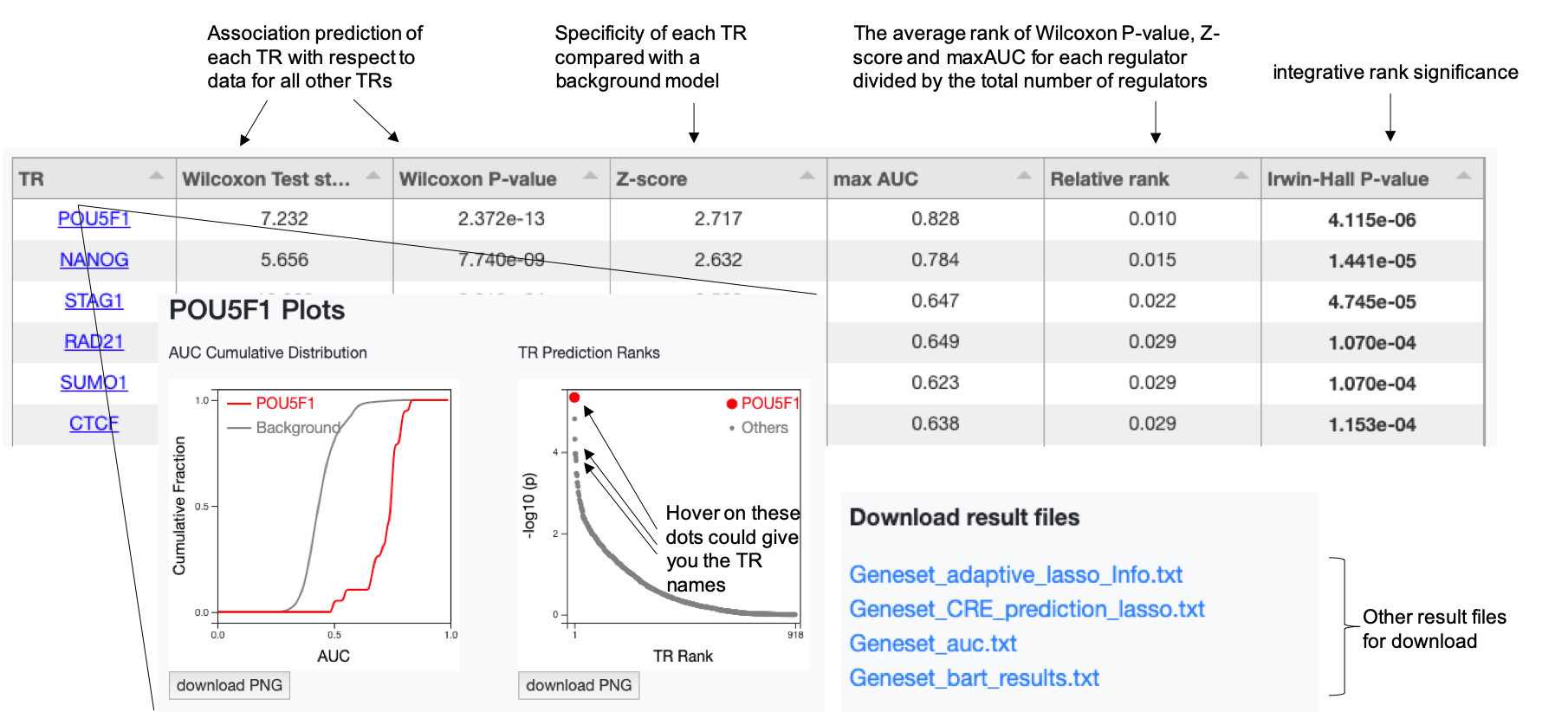
Output 1: Transcriptional regulator (TR) prediction table
We take this sub-table of a pre-run result from a gene set that were downregulated upon OCT4 (POU5F1) knocked down in a human embryonic stem cell as an example to illustrate. Another pre-run result from a ChIP-seq profile upon AR being enhanced in a human prostatic carcinoma cell line LNCaP is also provided as a reference for ChIP-seq data input.
- TR (Column 1): Transcriptional regulator name. When clicking a TR name, the analysis plots for this TR will be shown in a pop-up window.
- Wilcoxon test statistic & P-value (Columns 2 & 3): These two values indicate the level of association of each TR under the background of all other TRs. For each TR, we use Wilcoxon rank-sum test to compare the association scores from all ChIP-seq datasets for that regulator with the association scores from all ChIP-seq datasets for other TRs.
- Z-score (Column 4): This value is to assess the specificity of each TR compared with a background model. We build up background models using the Wilcoxon test statistics obtained from all annotated gene sets from the Molecular Signatures Database (MSigDB) for the Genelist mode or all H3K27ac ChIP-seq datasets from the data compendium for ChIP-seq data mode, respectively.
- Max AUC (Column 5): The maximum association score among multiple ChIP-seq datasets of that TR.
- Relative Rank (Column 6): The average rank of Wilcoxon test statistic, Z-score and maxAUC for each TR, divided by the total number of TRs.
- Irwin-Hall P-value (Column 7): This P-value indicates the integrative rank significance, using the Irwin-Hall distribution as the null distribution for unrelated ranks. One can use 0.01 as a threshold for significant TR identification.
Output 2: Data files and figure images
Besides the transcriptional regulator table, other output data files are also available for download, including the predicted genomic regulatory (cis-regulatory element) profile and analysis plots for each regulator. The image files are in high resolution with publication quality.
- *_adaptive_lasso_Info.txt provides regression information tells which representative H3K27ac samples are selected along with coefficients through adaptive lasso regression and sample annotations including cell line, cell type or tissue type. This is the output only generated in geneset mode.
- *_CRE_prediction_lasso.txt is the predicted cis-regulatory profile of the input gene set and is a ranked list of all CREs (UDHS) in the genome. The higher the score, the more likely the regulatory element regulates the input gene set. This is the output only generated in geneset mode.
- *_auc.txt provides the association score of each of the TR ChIP-seq dataset with the genome cis-regulatory profile.